Spatial growth-transform models
Michael Barkasi
August 4, 2026 (v1.0)
tutorial_SGT.RmdIntroduction
The simplest mathematical models of neural networks are built from homogeneous McCulloch-Pitts neurons with atemporal, scalar-weight connections between them. Biological neural networks, such as those in the mammalian brain, are far more complex. They contain different types of neurons each with their own electrical behavior and spatiotemporally extended connections.
A sample neural network diagram from the original 1943 paper by McCulloch and Pitts.
Computational neuroscientists standardly model biological neural networks as dynamical systems, i.e., as having time-dependent states. Growth-transform (GT) models, extended to spiking neural networks by Gangopadhyay and Chakrabartty, are one example. These models treat the change in membrane potential over time, \partial v/\partial t, as a growth-transform of the membrane potentials v on the net metabolic power \mathcal{H} of a network, in the sense that it’s assumed that \partial v/\partial t satisfies the Baum-Eagon inequality: \mathcal{H}(v)\leq \mathcal{H}(v + \partial v/\partial t) In other words: \frac{\partial\mathcal{H}}{\partial v}\frac{\partial v}{\partial t} \leq 0 Thus, GT models are essentially energy-minimization models of neural dynamics. The model parameters provided by DACx are based on a biological interpretation of the underlying mathematics of GT models.
In practical terms, instead of only a weight between two homogeneous neurons, GT models use both a cell-type-dependent transconductance parameter and a cell-type-dependent temporal modulation factor to determine network behavior. The transconductance parameter is essentially a traditional connection weight, while the temporal modulation factor allows for capturing the electrodynamics \partial v/\partial t of different neuron types at a single spatial point.
The GT models implemented in the DACx package add a third aspect of biological realism not captured by the original formulation of Gangopadhyay and Chakrabartty: a transmission velocity parameter for each neuron type. While the temporal modulation factor controls membrane voltage over time at a single spatial point, the transmission velocity parameter determines the rate at which changes in membrane voltage propagate between neurons. For this reason, we refer to our GT models as spatial GT (SGT) models.
SGT models thus allow for network topologies that not only capture connection strengths between neurons, but also the different types of neurons and their electrodynamics across both time and space. A separate tutorial demonstrates how to build spatially extended network topologies for these models from circuit motifs. This tutorial explains SGT models in more detail.
Network nodes
Let’s set up the R environment by clearing the workspace, setting a random-number generator seed, and loading the DACx package.
# Clear the R workspace to start fresh
rm(list = ls())
# Set seed for reproducibility
set.seed(12345)
# Load DACx package
library(DACx, quietly = TRUE) Next, we create a new network object with the new.network function.
network.node <- new.network()The object initialized by new.network is a single-node network. As we use the term, a “node” is not necessarily a single neuron, but is rather a cluster of nearby neurons with local recurrent connections. These nodes are expected to be (approximately) fully connected, with cells of each type synapsing into cells of all other types. For this tutorial, our nodes will include two distinct neuron types: an excitatory type, layer 4 principal neurons (spiny stellates), and an inhibitory type, somatostatin (PV) interneurons.
Nodes are defined by their constitutive cell types and node size (i.e., expected number of neurons per type). DACx comes with a number of preloaded cell types and functions for modifying existing cell types and adding new ones. Details about cell types are discussed in another tutorial. Although many of the values are just the defaults, we will use the modify.cell.type to explicit set all values, for later reproducibility.1 The details of these parameters are explained in the tutorial on cell types.
For now, it suffices to note that PV interneurons are highly responsive cells with a high rate of fire, little adaptation (short-term depression), but also little memory – that is, they have a high leak current and don’t integrate signals. Thus, they function as coincidence detectors (integrating only near-simultaneous input spikes) and send strong inhibitory signals. Conversely, spiny stellate cells are slower to respond, have higher adaptation, but more memory. They serve as signal integrators, integrating input spikes over longer time stretches.
modify.cell.type(
"PV",
# Synaptic transmission
valence = -1,
synaptic_conductance = list( # nS
spiny_stellate = 0.25,
PV = 1.5),
# Membrane kinetics
leak_conductance = 10.0, # nS, High membrane conductance for fast kinetics
tau_fast = 0.1, # ms, Short for fast responses
tau_slow = 60.0, # ms
tau_Vs = 10.0, # ms/spike, Fast recovery for little adaptation
I_slow = 0.02, # concentration/spike
U_Vs = 0.05, # concentration/spike
max_spike_rate = 0.5, # spikes/ms
# Intercell transmission
transmission_velocity = 20e3, # micons/ms
spine_density = 0.0,
axon_target = "soma",
# Membrane potential and spiking
I_spike = 1e3, # pA
spike_potential = 35, # mV
resting_potential = -70, # mV
threshold = -50, # mV
# Neurite structure
axon_branch_count = 10,
dendrite_branch_count = 10,
branch_independence = 0.625,
branch_spread = 0.625,
# Apical dendrite parameters
apical_target_layer = "none"
)
modify.cell.type(
"spiny_stellate",
# Synaptic transmission
valence = 1,
synaptic_conductance = list( # nS
PV = 1.0,
spiny_stellate = 0.2),
# Membrane kinetics
leak_conductance = 2.5, # nS, Low conductance for slow kinetics
tau_fast = 2.0, # ms, Long for slow responses
tau_slow = 60.0, # ms
tau_Vs = 100.0, # ms/spike, Slow recover for more adaptation
I_slow = 0.01, # concentration/spike
U_Vs = 0.1, # concentration/spike
max_spike_rate = 0.1, # spikes/ms
# Intercell transmission
transmission_velocity = 10e3, # microns/ms, slower transmission than PV cells
spine_density = 0.5,
axon_target = "spine",
# Membrane potential and spiking
I_spike = 1e3, # pA
spike_potential = 35, # mV
resting_potential = -70, # mV
threshold = -55, # mV
# Neurite structure
axon_branch_count = 10,
dendrite_branch_count = 10,
branch_independence = 0.75,
branch_spread = 0.75,
# Apical dendrite parameters
apical_target_layer = "none"
)Mimicking the structure of the brain, nodes are arrayed into layers and columns, cortical and subcortical regions, and hemispheres. The network-topology tutorial explains how to set up this multi-node structure. For now, it suffices to note that this structure is set with the set.network.structure function. We will set up a single-node network, with an expected count of 50 for the spiny stellates and 5 for the PV interneurons.
network.node <- set.network.structure(
network.node,
neuron_types = c("spiny_stellate", "PV"),
neurons_per_node = c(50, 5),
synaptic_neighborhood = 30
)By default, the set.network.structure function creates no subcortical layers and sets the number of cortical layers, columns, patches (a secondary columnar axis perpendicular to the laminar axis), and hemispheres each to one. That is, it makes a single node. It also initializes local recurrent connections within that node. The node we just created can be visualized with the plot.network function. To keep the plot clean, we can use the arbor_density argument, which controls the proportion of cells for which the generated arbors are shown.
plt <- plot.network(network.node, arbor_density = 0.1)
plt$plot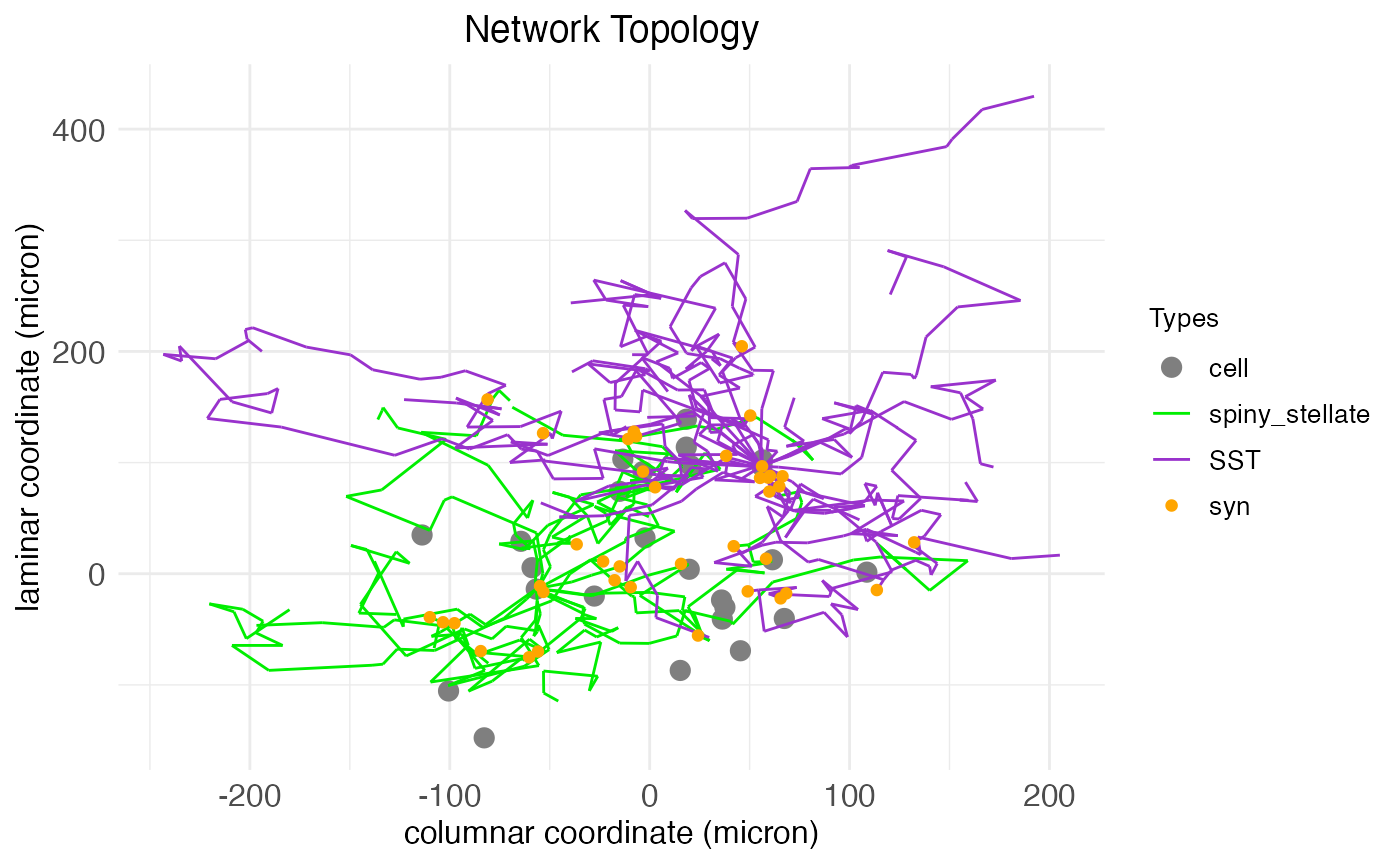
Here we see arbors for two cells, a spiny stellate and a PV interneuron. These arbors are generated via a biased random walk, the biasing factors fixed by cell type. The arbors include both axons and dendrites, which can be visualized explicitly by changing the arbor coloring. Although cells and arbors to-be-plotted are selected randomly, the plot.network function returns masks for the cells and arbors plotted, and can take this information to re-plot the same data again under different settings, e.g., coloring by arbor type instead of by cell type:
plt <- plot.network(
network.node,
arbor_density = 0.1,
soma_mask = plt$soma_mask,
arbor_idx = plt$arbor_idx,
edge_color = "is_axon"
)
plt$plot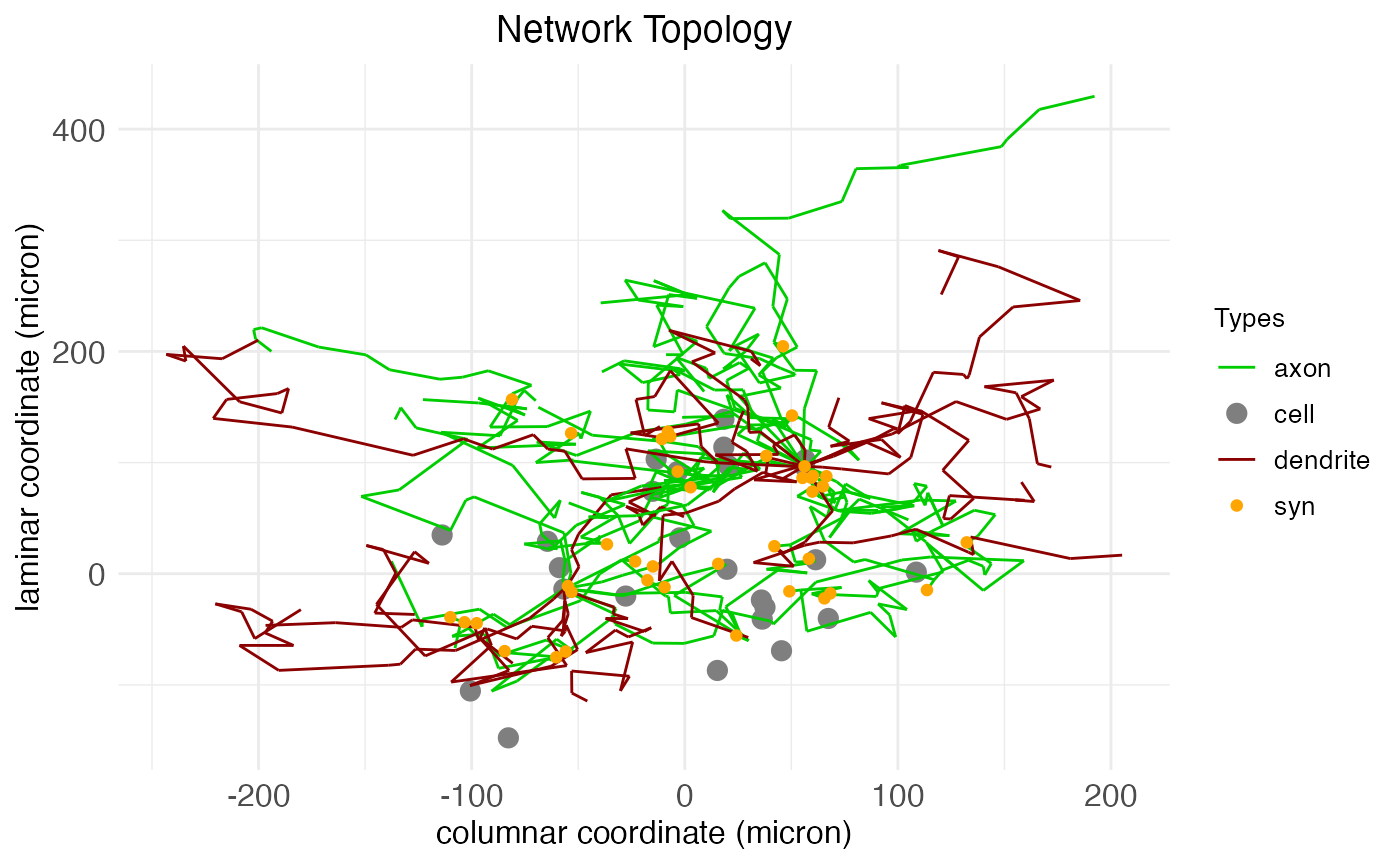
The existence and number of connections between cells – synapses, colored orange – are determined by the proximity of axons to dendrites. After the arbors are created, a separate algorithm looks for axon nodes within a certain small neighborhood of dendrite nodes and, if one is found, extends the axon to connect with the dendrite.
As the axis labels indicate, SGT models assign to each neuron a spatial coordinate giving its location along the laminar and columnar axes. While the above are 2D plots, there is in fact a third dimension, the “patch” dimension, which serves as a secondary columnar axis. All coordinates are continuous and real-valued and are used in conjunction with the transmission velocity parameter to simulate spike propagation over the axonal arbors. Here, for example, we can plot a 3D representation of our node including all arbors, colored by cell type:
plt <- plot.network(
network.node,
arbor_density = 1.0,
threedim = TRUE
)
plt$plotAs can be seen, even for a small single node, the arborization and number of synapses can be extensive. We can quantify the extent by calling the fetch.network.components function, which returns a list of all components of the network, including a print out of summary data:
plot.network(
network.node,
arbor_density = 1.0,
reconstruct_arbors = FALSE
)$plot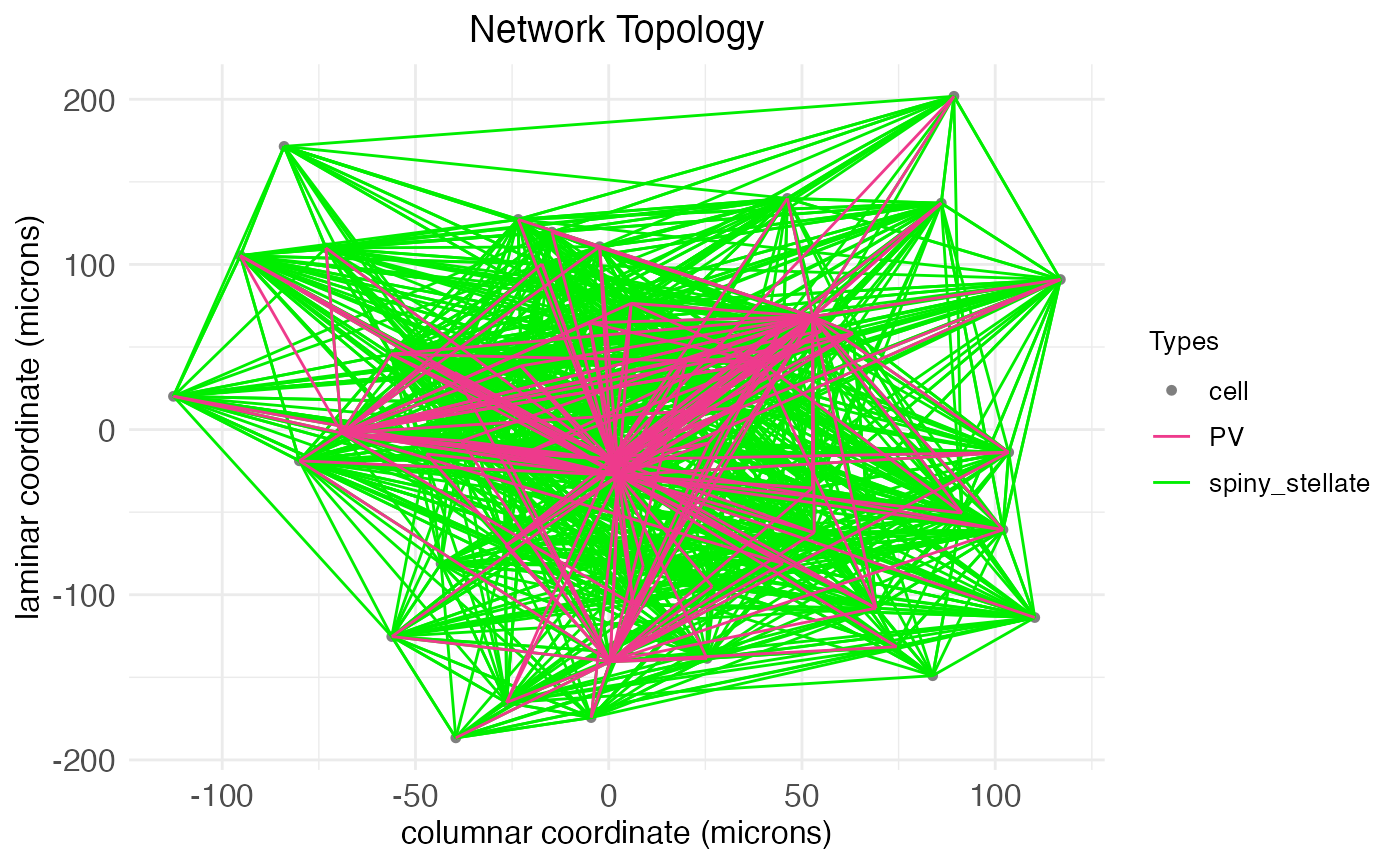
ntw <- fetch.network.components(network.node, include_arbors = TRUE)## Summary of network:
## Number of neurons: 55
## Number of synapses: 1563
## Hemisphere names: left
## Number of hemispheres: 1
## Subortical layer names:
## Number of subcortical layers: 0
## Cortical layer names: layer
## Number of cortical layers: 1
## Number of columns: 1
## Number of patches: 1
## Cell types used: spiny_stellate, PV
## Motifs used: local connectionsAs the axis labels in the previous plots also indicate, there is a physically meaningful unit attached to the dimensions: microns. The formulas used to compute spatial coordinates are discussed in the network-topology tutorial.
SGT simulations
The function run.SGT runs a simulation of spiking activity across a network using a SGT model. The function is just a wrapper over the SGT method of C++ network objects. It takes five arguments:
- network: A network created by the new.network function and structured by the set.network.structure function.
- stimulus_current_matrix: A matrix of input currents (in mA) over the duration of the simulation, rows representing neurons and columns representing time bins.
- dt: Time-step size for simulation, in ms. Default is 10^{-3}.
- initial_potential: Initial value for membrane potential, applied to all cells. Default is -70 mV.
- return_v_only: Boolean specifying whether to return only the membrane potential traces and related spike counts. If FALSE, will also return matrices containing traces related to the temporal modulation term. Default is TRUE.
The number of columns of stimulus_current_matrix determines the length of the simulation. In essence, the function run.SGT answers the question: How would the network respond to this stimulus current over this amount of time?
For example, let’s create a 500ms simulation for the node we created above. From the above call to fetch.network.components, we know there are 15 neurons in our network. We can load this value directly from the function output:
n_neurons <- ntw$n_neuronsThis gives us the number of rows needed in our stimulus current matrix. For the number of columns, we need to know the number of time steps required:
stim_time_ms <- 500
dt <- 1e-3
n_steps <- stim_time_ms/dt
cat("Number of time steps in the simulation:", n_steps)## Number of time steps in the simulation: 5e+05Now, suppose we want our simulation to involve a 200ms input current to just the spiny stellates, starting at 100ms. We can compute the initial and final time steps of this current, plus a mask for the spiny stellates, as follows:
# Set stimulus start and length
stim_length_ms <- 300
stim_start_ms <- 100
# Find start and end steps of the input stimulus current
stim_length <- stim_length_ms / dt
stim_start <- stim_start_ms / dt
stim_end <- stim_start + stim_length - 1
# Find mask for principal neurons
spiny_stellate_mask <- ntw$neuron_type_name == "spiny_stellate"A final question is how much current to apply. For this simulation, we’ll use a constant current of 100 pico amp to the spiny stellates during the stimulus period.
stimulus_current_matrix <- matrix(0, nrow = n_neurons, ncol = n_steps)
stimulus_current_matrix[spiny_stellate_mask, stim_start:stim_end] <- 150With the stimulus current matrix in hand, we can run the simulation:
sim_results <- run.SGT(
network.node,
stimulus_current_matrix,
dt
)The result of the function run.SGT is a matrix of spike traces formatted similar to stimulus_current_matrix: each row represents a neuron and each column represents a time step from the simulation. Each entry is the membrane potential of the neuron at that time bin, in mV. The order of neurons and time steps matches across the input stimulus-current and output spike-trace matrices, of course. In addition, a vector of spike counts for each neuron (giving the number of times each neuron spiked) in the network is also returned. Both are returned in a list of two elements, sim_traces and spike_counts.
We can view the head of the simulation traces:
print(sim_results$v_traces[1:10,1:10])## [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8] [,9] [,10]
## [1,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [2,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [3,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [4,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [5,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [6,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [7,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [8,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [9,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70
## [10,] -70 -70 -70 -70 -70 -70 -70 -70 -70 -70As well as the head of the spike counts:
## [1] 9 9 9 9 9 10The neurons package also includes the function plot.network.traces, which takes a network object with a trace matrix and produces a plot of the traces, putting all neurons of the same type together.
plot.network.traces(network.node, input_matrix = stimulus_current_matrix)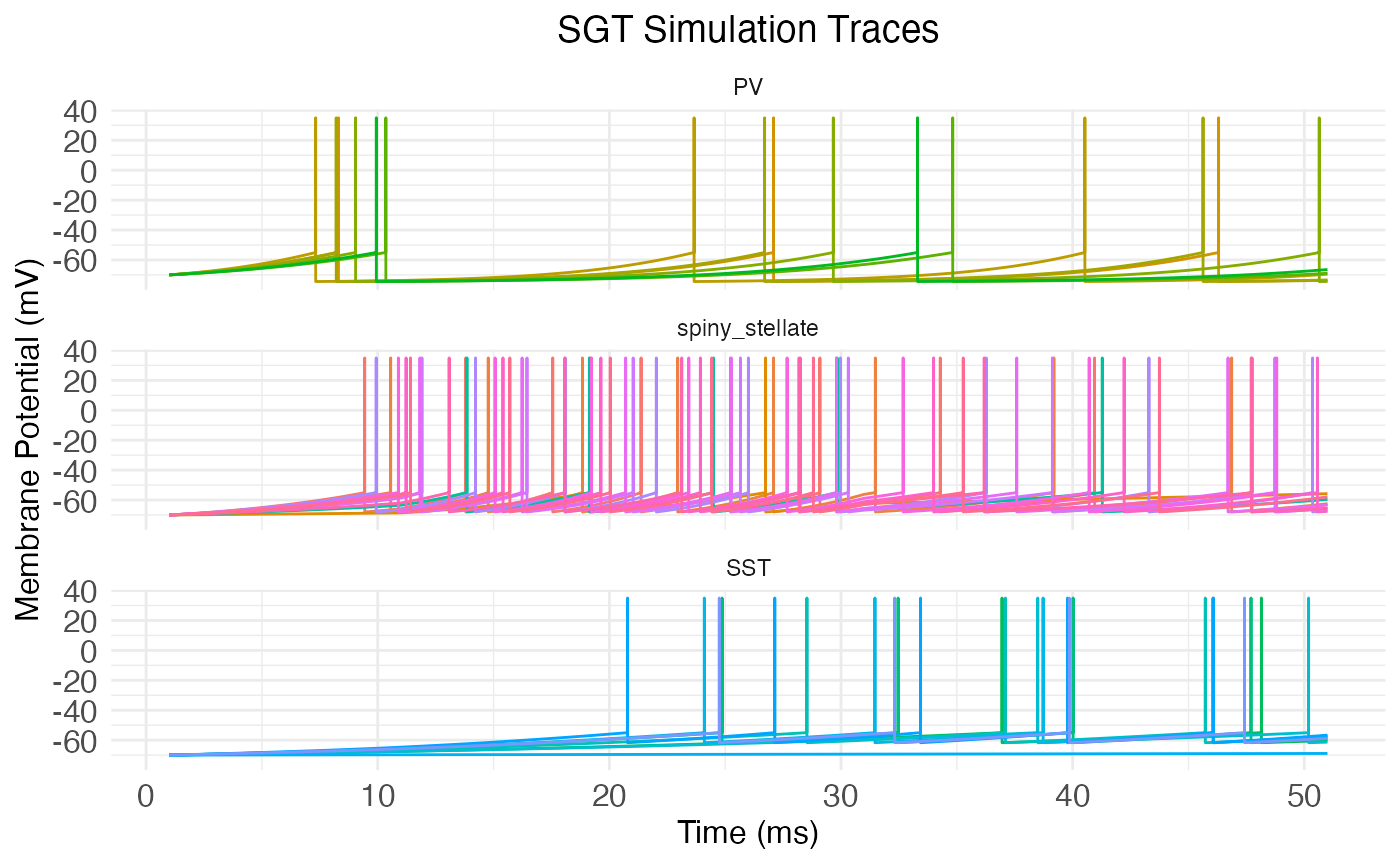
Spatial lag
What about I_\mathrm{synaptic\;transmission}, the input current induced by synaptic transmission across all synapses? We assume that the induced post-synaptic current is equal to the inducing pre-synaptic potential, modulated by the synaptic conduction. However, the complicating factor is that, given spatial distance between cells and synaptic transmission time, the relevant pre-synaptic potential v at time t from pre-synaptic neuron N may not be v(t), but rather v(t^\prime) for some t^\prime < t.
Let \vec{v} = \langle v_1, v_2, \ldots, v_n\rangle be the vector of membrane potentials for all n neurons in the network at time t. Further, let V be a n\times n time-dependent matrix which captures how each neuron “sees” the others. Specifically, V_{ij}(t) is the membrane potential of neuron N_i that reaches neuron N_j at time t. Then, assuming Q is an n\times n matrix of synaptic connections such that Q_{ij} is the conductance from neuron j to neuron i, we have that: I_\mathrm{synaptic\;transmission}(N_i) = \sum_{j=1}^n Q_{ij}V_{ji}(t)